Troubleshooting Liquid/Liquid Extraction
Course Details
Quick Guide
Author:  | 0h 15m
| 0h 15m
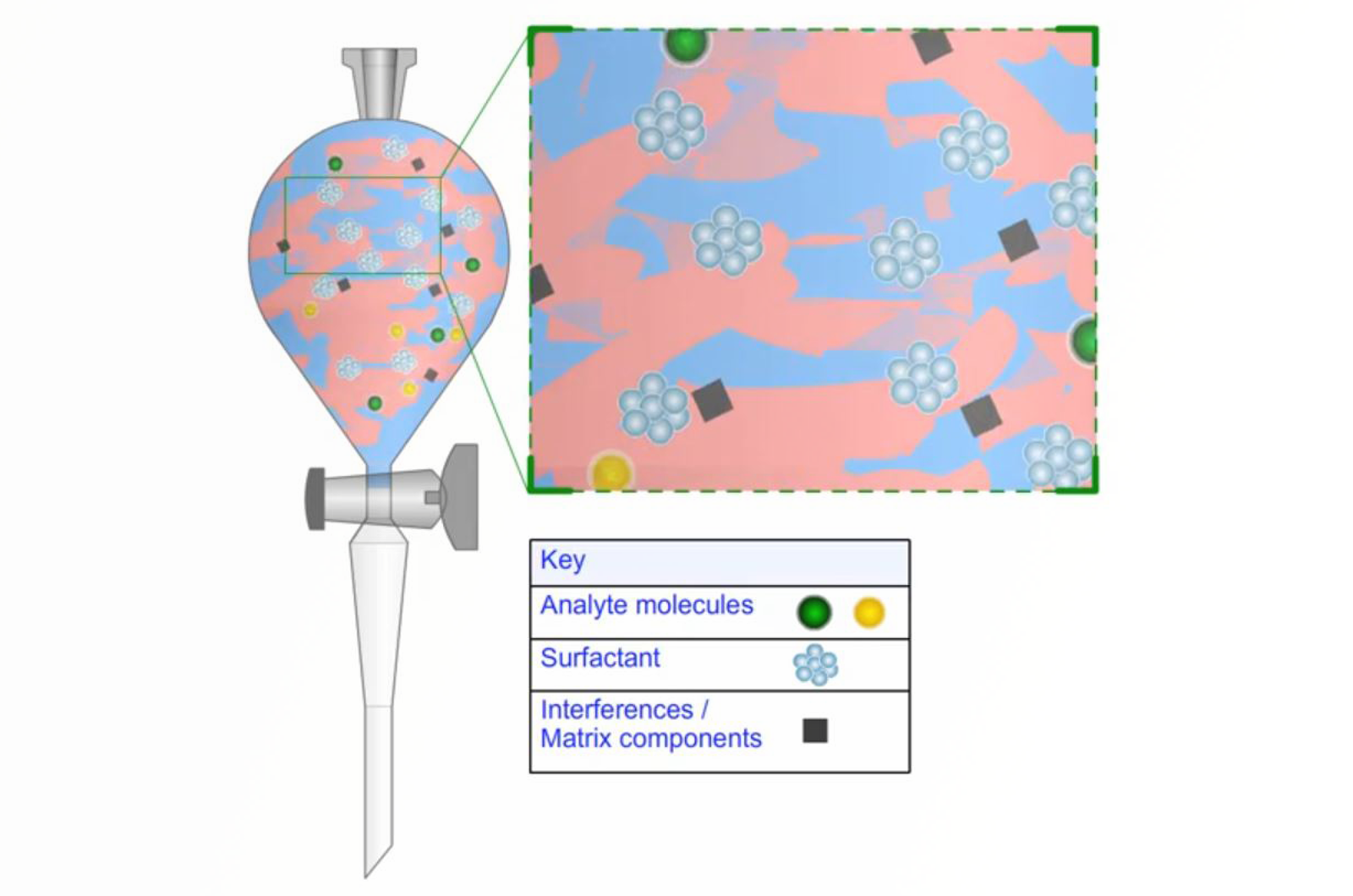
Liquid/liquid extraction (LLE) is the most widely used extraction technique for liquid samples. The information provided in this article will mainly focus on liquid-liquid extraction (LLE) using a separatory funnel, however, many of the principles are applicable to the more modern miniaturized approaches to LLE i.e. single-drop microextraction (SDME), flow-injection extraction (FIE), membrane extraction techniques, and supported liquid extraction (SLE).