Introduction to Mass Spectrometry for Biomolecules
Endorsed by

Course Details
Module
Premier
Author:  | 0h 30m
| 0h 30m
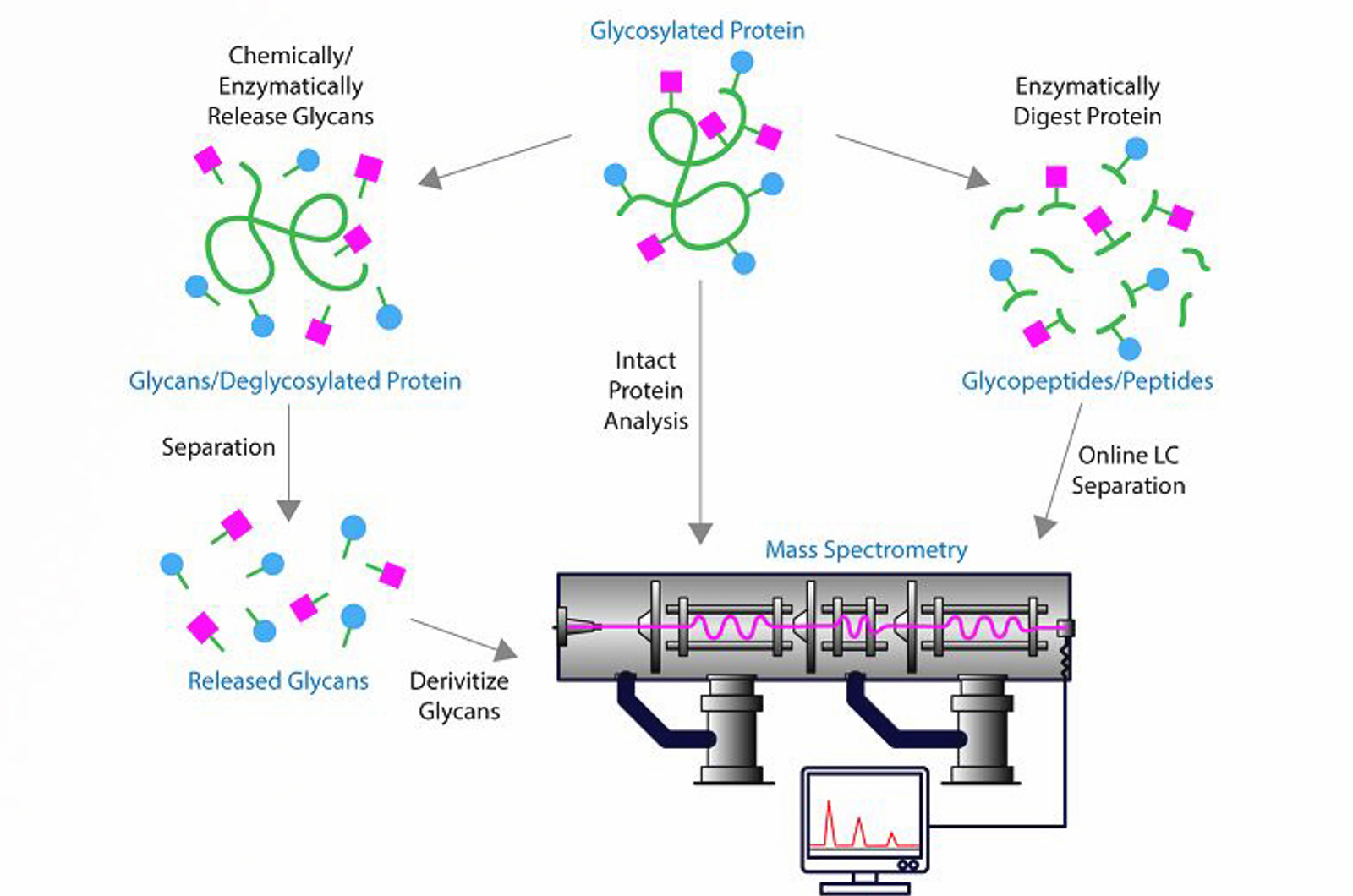
Mass spectrometry (MS) plays an important role in the analysis and characterization of protein biopharmaceuticals. It is utilized at almost every stage of development, including clone selection, cell culture process development, purification process development, formulation development, stability studies, comparability studies, structural characterization and quality control. This module provides an introduction to mass spectrometry for biomolecules.
Topics include:
- Intact protein and middle-up measurements
- Top-down and middle-down measurements
- Bottom-up measurements
- Glycan measurements